Choroba Creutzfeldta-Jakoba jest rzadką chorobą neurodegeneracyjną, która gwałtownie, progresywnie i poważnie atakuje mózg. Zdecydowanie warto zwrócić na nią uwagę, ponieważ jej dynamika działania jest niezwykle niebezpieczna.
CJD stopniowo niszczy komórki mózgu, prowadząc do powstawania małych dziur w mózgu. Osoby z CJD mogą doświadczać ataksji, co oznacza trudności w kontrolowaniu ruchów ciała, a także nieprawidłowego chodu, zaburzeń mowy i demencji.
Niestety, choroba ta jest zawsze śmiertelna, a aktualnie nie ma skutecznego lekarstwa.
Globalnie, CJD dotyka rocznie około miliona osób, w tym także w Stanach Zjednoczonych.
Przyczyny mogą być sporadyczne, dziedziczne lub nabyte. W większości przypadków choroba występuje u osób powyżej 60. roku życia, podczas gdy jej występowanie u osób poniżej 30. roku życia jest niezwykle rzadkie.
Osoba z CJD prawdopodobnie nie przetrwa dłużej niż rok po wystąpieniu pierwszych objawów.
Czym jest CJD?

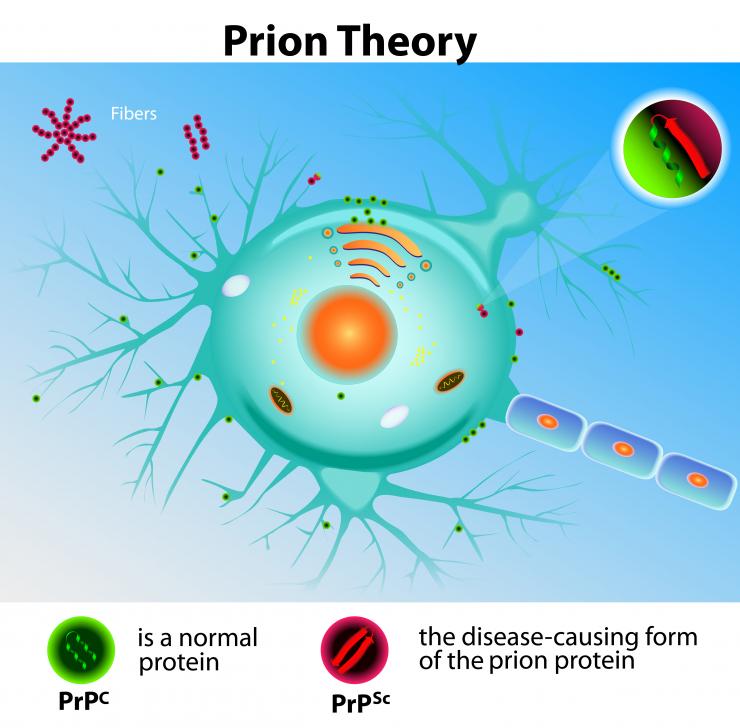
CJD jest pasażowalną encefalopatią gąbczastą (TSE), która w miarę upływu czasu poważnie uszkadza mózg. Powstaje w wyniku działania czynnika zakaźnego, znanego jako prion. Prion to nie wirus ani bakteria, lecz nieprawidłowa forma białka, która wywołuje szereg problemów zdrowotnych.
Inne typy TSE obejmują zespół Gerstmanna-Sträusslera-Scheinkera (GSS), śmiertelną rodzinną bezsenność oraz kuru. Przykłady obejmują również trzęsawki u owiec i kóz oraz bydlęcą encefalopatię gąbczastą (BSE), znaną jako «choroba szalonych krów».
Podobne encefalopatie występują u innych gatunków i udowodniono, że mogą one być przenoszone na zwierzęta laboratoryjne.
Centrum Kontroli i Zapobiegania Chorób (CDC) wskazuje, że klasyczny CJD nie jest powiązany z BSE ani innymi wariantami tej choroby.
Objawy
CJD ma długi okres inkubacji, a objawy mogą pojawić się nawet po 40 latach. Gdy zaczynają się objawy, stan pacjenta szybko się pogarsza, a większość ludzi umiera w ciągu roku.
Charakterystyczne objawy CJD to szybki postęp demencji oraz mioklonie, które są mimowolnymi skurczami mięśni. Można zauważyć zmiany nastroju, zachowania, osobowości, utratę pamięci oraz problemy z oceną sytuacji. Choroba może przypominać demencję Alzheimera lub chorobę Huntingtona, ale w przypadku CJD objawy ewoluują w ciągu dni, a nie lat.
W miarę postępu choroby problemy z koordynacją oraz mioklonie nasilają się, a wzrok staje się osłabiony, co w końcu prowadzi do ślepoty. Ostatecznie pacjent traci zdolność poruszania się i mówienia, a jego stan przechodzi w śpiączkę.
Sekcje zwłok tkanki mózgowej ujawniły, że CJD wiąże się z pewnymi unikalnymi zmianami, które nie są spotykane w innych formach demencji.
Istnieje kilka wariantów CJD, które niekoniecznie są związane z klasycznym CJD, a objawy oraz przebieg choroby mogą być zróżnicowane.
Przyczyny
CJD występuje, gdy białko prionowe, nieprawidłowa forma białka amyloidu, powoduje nieprawidłowości w innych białkach. Nagromadzenie i zniekształcenie prionów w komórkach mózgowych prowadzi do uszkodzenia mózgu i śmierci.
Może mieć charakter sporadyczny, dziedziczony lub nabyty.
Sporadyczne CJD
W 85% przypadków CJD występuje w formie sporadycznej, bez wyraźnych czynników ryzyka.
Odziedziczony CJD
Od 5% do 10% przypadków jest dziedziczonych. Dzieje się tak, gdy dochodzi do zmiany w genie, który kontroluje tworzenie białek prionowych. Może występować w rodzinach z historią CJD, a mutacja może występować w komórkach jajowych lub plemnikach, co zwiększa ryzyko rozwoju choroby u potomstwa.
Priony nie mają informacji genetycznej i nie potrzebują genów do rozmnażania, ale mutacja w genie normalnego białka prionowego organizmu może powodować nienormalne działanie prionów.
Zidentyfikowano kilka różnych mutacji w genie prionu. Szczególna mutacja występująca w każdej rodzinie wpływa na to, jak często choroba pojawia się i jakie objawy są najbardziej zauważalne.
Nie każda osoba z mutacjami w genie białka prionowego rozwija CJD.
Nabyte CJD
Nie ma dowodów na to, że jakikolwiek rodzaj CJD może być przenoszony z jednej osoby na drugą, ale pewne procedury medyczne zostały powiązane z transmisją CJD.
Procedury te obejmują:
- przeszczep rogówki
- implanty elektrody
- przeszczep opony twardej
- stosowanie ludzkiego hormonu wzrostu
Około 1% przypadków jest przenoszonych przez znane lub wysoce podejrzane narażenie na tkankę mózgu lub układu nerwowego.
Gąbczasta encefalopatia bydła
W latach 90-tych jeden typ CJD był powiązany z ekspozycją na BSE, która występuje u bydła.
Uważano, że transmisja była związana ze spożywaniem żywności. Ten wariant miał tendencję do wpływania na młodszych pacjentów i trwał dłużej.
BSE dotyka wiele gatunków, w tym bydło, ludzi i koty.
Niektórzy naukowcy uważają, że niezwykły «wolny wirus» lub inny organizm może powodować CJD, ale do tej pory nie wyizolowano konkretnego wirusa lub organizmu u osób z tą chorobą.
Czynnik powodujący CJD ma cechy, które nie są typowe dla wirusów i bakterii.
Należą do nich długi okres inkubacji, trudność w zniszczeniu oraz brak jakiejkolwiek informacji genetycznej w postaci kwasów nukleinowych, takich jak DNA czy RNA.
Naukowcy są zdania, że CJD oraz inne TSE są wywoływane nie przez żywy organizm, lecz przez priony. Priony nie są żywe, lecz są białkami o nienormalnych strukturach, które rozprzestrzeniają się w mózgu.
Ta ekspansja prowadzi do uszkodzenia tkanki mózgowej i powoduje charakterystyczne objawy CJD.
Diagnoza
Nie ma testu, który potwierdziłby diagnozę CJD. Tylko biopsja mózgu może to zrobić, co jest zbyt ryzykowne, gdy pacjent jest jeszcze żywy.
Testy mogą pomóc w ustaleniu najbardziej prawdopodobnej przyczyny objawów.
Fizyczne badanie obejmuje ocenę skurczów mięśni i odruchów pacjenta, które mogą być bardziej reaktywne niż normalnie. Mięśnie mogą być nadmiernie napięte lub osłabione, w zależności od tego, gdzie choroba wpływa na mózg.
Badania wzrokowe mogą wykryć częściową ślepotę, której pacjent wcześniej nie zauważył.
Elektroencefalogram (EEG) może ujawnić nieprawidłowe impulsy elektryczne.
Tomografia komputerowa (TK) lub rezonans magnetyczny (MRI) mogą wykluczyć udar jako przyczynę objawów.
Nakłucie lędźwiowe może zbadać płyn rdzeniowy, aby wykluczyć inne przyczyny demencji. Może ujawnić infekcję lub zwiększone ciśnienie w ośrodkowym układzie nerwowym.
Jeśli w płynie zostanie wykryte białko 14-3-3, a pacjent wykazuje typowe objawy, istnieje duża szansa, że ma CJD.
Biopsje mózgu po śmierci ujawniają, że tkanka mózgowa jest gąbczasta, z widocznymi małymi otworami w miejscach, gdzie zniszczeniu uległy kępki komórek nerwowych.
Leczenie
Nie ma lekarstwa na CJD, a żadne leki nie pomagają w kontrolowaniu lub spowolnieniu postępu choroby.
Leczenie koncentruje się na łagodzeniu objawów i zapewnieniu pacjentowi jak największej wygody.
Leki przeciwbólowe mogą pomóc w łagodzeniu bólu. Klonazepam i walproinian sodu mogą być pomocne w redukcji mimowolnych ruchów, takich jak skurcze mięśni.
W późniejszych etapach pacjent często wymaga przenoszenia, aby zapobiec odleżynom. Cewnik może być używany do drenażu moczu, a karmienie może odbywać się za pomocą płynów dożylnych.
Zapobieganie
Środki zapobiegawcze obejmują sterylizację całego sprzętu medycznego, aby zabić wszelkie organizmy, które mogą wywołać chorobę oraz unikanie przyjmowania darowizn rogówki od osób z rozpoznaną lub podejrzewaną historią CJD.
W większości krajów obowiązują surowe wytyczne dotyczące zarządzania zakażonymi krowami oraz ograniczenia dotyczące pasz, aby uniknąć ryzyka przenoszenia innych form TSE na ludzi.
Osoby narażone na kontakt z osobami z rozpoznaniem CJD powinny przestrzegać pewnych wytycznych.
Wytyczne te obejmują:
- zabezpieczenie otwartych ran, skaleczeń i otarć na skórze
- noszenie rękawiczek podczas pracy z tkankami pacjenta, krwią lub płynami
- stosowanie jednorazowych fartuchów lub odzieży podczas kontaktu z pacjentem
- używanie ochrony twarzy, ochrony oczu lub masek, gdy istnieje ryzyko rozpryskania skażonego płynu
- sterylizację sprzętu używanego na lub w pobliżu pacjenta
Wciąż prowadzone są badania nad rolą prionów w CJD, aby zrozumieć, w jaki sposób choroba wpływa na mózg oraz znaleźć skuteczne metody leczenia.
Aktualne Badania i Statystyki na Rok 2024
W 2024 roku badania nad chorobą Creutzfeldta-Jakoba koncentrują się na lepszym zrozumieniu mechanizmów prionów oraz ich interakcji z komórkami mózgowymi. Ostatnie wyniki wskazują na potencjalne terapie oparte na molekułach, które mogłyby hamować zjawisko agregacji prionów.
Statystyki z ostatnich lat pokazują, że choć CJD pozostaje rzadką chorobą, to liczba przypadków wzrasta, co może być wynikiem lepszej diagnostyki oraz zwiększonej świadomości wśród lekarzy. W 2023 roku odnotowano około 1,2 miliona przypadków na całym świecie, co stanowi wzrost o 5% w porównaniu do roku poprzedniego.
Nowe badania kliniczne prowadzone są również w celu oceny skuteczności różnych strategii leczenia objawowego, takich jak terapia zajęciowa oraz wsparcie psychologiczne, co ma na celu poprawę jakości życia pacjentów oraz ich rodzin.
Eksperci podkreślają znaczenie wczesnego rozpoznawania objawów oraz edukacji społeczeństwa w zakresie tej choroby, co może przyczynić się do lepszego zarządzania przypadkami CJD w przyszłości.