Stwardnienie zanikowe boczne, znane jako ALS, to rodzaj choroby neuronu ruchowego. Dotyczy grupy postępujących schorzeń neurologicznych, które prowadzą do dysfunkcji nerwów odpowiedzialnych za ruchy mięśni.
Choroba ta skutkuje osłabieniem mięśni oraz zmianami w funkcjonowaniu organizmu. W zaawansowanych stadiach ALS wpływa na nerwy kontrolujące oddychanie, co może prowadzić do poważnych konsekwencji zdrowotnych.
ALS jest najczęściej występującym rodzajem choroby neuronu ruchowego (MND). Często określa się ją mianem choroby Lou Gehriga, nawiązując do słynnego baseballisty, który zmagał się z tym stanem. Wydarzenie «Ice Bucket Challenge» w 2014 roku miało na celu zwiększenie świadomości oraz funduszy na badania nad tą chorobą.
Centrum Kontroli i Prewencji Chorób (CDC) szacuje, że w 2016 roku w Stanach Zjednoczonych było od 14 500 do 15 000 przypadków ALS. Co roku około 5000 osób otrzymuje diagnozę. Globalnie uważa się, że choroba dotyka od 2 do 5 osób na każde 100 000 mieszkańców.
Większość pacjentów z ALS żyje od 3 do 5 lat po wystąpieniu pierwszych objawów, jednak około 10% z nich może przeżyć 10 lat lub dłużej.
Obecnie nie ma lekarstwa na ALS, ale dostępne terapie mogą złagodzić objawy i poprawić jakość życia pacjentów.
Szybkie fakty na temat ALS
Oto kilka kluczowych punktów dotyczących ALS. Więcej szczegółów znajduje się w głównym artykule.
- ALS wpływa na komórki nerwowe w mózgu i rdzeniu kręgowym, co prowadzi do osłabienia mięśni, utraty funkcji motorycznych, paraliżu, problemów z oddychaniem i, ostatecznie, śmierci.
- Większość osób z ALS będzie żyła od 3 do 5 lat po pojawieniu się objawów.
- Dokładna przyczyna nie jest znana, ale mogą być w nią zaangażowane czynniki środowiskowe i genetyczne.
- Obecnie nie ma lekarstwa, a leczenie ma na celu złagodzenie objawów, zapewnienie wsparcia społecznego i emocjonalnego oraz prawdopodobnie spowolnienie postępu choroby.
Co to jest ALS?

ALS to rodzaj MND, który atakuje komórki nerwowe odpowiedzialne za dobrowolne ruchy mięśni. Są to działania, które można kontrolować, takie jak ruchy ramion, twarzy i nóg.
Neurony motoryczne znajdują się w mózgu i rdzeniu kręgowym. W miarę postępu ALS, komórki te degenerują i umierają, co prowadzi do utraty zdolności wysyłania sygnałów do mięśni. W rezultacie mózg przestaje kontrolować ruchy dobrowolne, a mięśnie stają się słabe i zanikają.
W miarę zaawansowania choroby, ALS wpływa na wszystkie dobrowolne mięśnie. Osoby dotknięte chorobą nie mogą już kontrolować swoich ramion, twarzy ani nóg. Z czasem niemożność oddychania bez wsparcia może prowadzić do niewydolności oddechowej.
Połowa pacjentów z ALS przeżywa 3 lata lub dłużej po postawieniu diagnozy, ale niektórzy żyją znacznie dłużej. Około 20% osób przeżywa 5 lat lub więcej, 10% żyje 10 lat lub dłużej, a 5% może przeżyć 20 lat.
Steven Hawking, znany fizyk, został zdiagnozowany z ALS w wieku 21 lat. Dziś, mając ponad 70 lat, pozostaje liderem w dziedzinie nauki.
Przyczyny i rodzaje ALS
Przyczyny ALS nie są w pełni zrozumiane. Istnieją różne rodzaje tej choroby, które różnią się objawami oraz występowaniem genetycznym.
ALS może być sporadyczny lub rodzinny.
Sporadyczny ALS występuje przypadkowo i stanowi od 90 do 95% wszystkich przypadków. Nie ma wyraźnego czynnika ryzyka ani przyczyny.
Rodzinny ALS jest dziedziczony i stanowi około 5 do 10% przypadków. Dziecko osoby z ALS ma 50% szans na rozwinięcie tej choroby. Rzadko zdarza się, że choroba występuje u osób w wieku dojrzewania. Naukowcy badają, które geny mogą być w to zaangażowane.
Inne możliwe przyczyny ALS to:
- Zdezorganizowana odpowiedź immunologiczna: Układ odpornościowy może atakować niektóre komórki organizmu, prowadząc do uszkodzenia komórek nerwowych.
- Nierównowaga chemiczna: osoby z ALS często mają wyższy poziom glutaminianu, chemicznego przekaźnika w mózgu, co może być toksyczne dla neuronów.
- Niewłaściwe przetwarzanie białek: jeśli białka nie są właściwie przetwarzane przez komórki nerwowe, mogą gromadzić się w nieprawidłowy sposób, co prowadzi do śmierci komórek nerwowych.
Możliwe czynniki środowiskowe
Czynniki środowiskowe mogą również odgrywać istotną rolę w rozwoju ALS.
Jedno z badań wykazało, że personel wojskowy, który służył w rejonie Zatoki podczas wojny w 1991 roku, miał wyższe ryzyko rozwoju ALS niż personel wojskowy rozmieszczony w innych miejscach.
Zidentyfikowano kilka możliwych powiązań między ALS a ekspozycją na:
- urazy mechaniczne lub elektryczne
- służbę wojskową
- wysoki poziom aktywności fizycznej
- wysoki poziom ekspozycji na chemikalia rolnicze
- wysokie stężenia różnych metali ciężkich
Nie ma jednak jednoznacznych dowodów na to, że konkretne zmiany stylu życia mogą zmniejszyć ryzyko wystąpienia ALS.
Symptomy i objawy
Objawy ALS zazwyczaj pojawiają się, gdy pacjent ma 50-60 lat, choć mogą występować również w innych grupach wiekowych.
Progresja choroby jest różna w zależności od osoby. We wczesnych stadiach objawy mogą być ledwo zauważalne, ale osłabienie staje się bardziej zauważalne w miarę upływu czasu.
Typowe objawy to:
- trudności w wykonywaniu codziennych czynności, w tym chodzenia
- zwiększona niezdarność
- osłabienie kończyn, dłoni i stóp
- skurcze oraz drganie rąk, nóg lub języka
- trudności w utrzymaniu prawidłowej postawy oraz głowy w górze
- niekontrolowane wybuchy śmiechu lub płaczu, znane jako labilność emocjonalna
- zmiany poznawcze
- brzęczenie mowy oraz trudności z artykulacją
- ból
- zmęczenie
- problemy z śliną i śluzem
- trudności w oddychaniu i połykaniu w późniejszych stadiach
Progresywne osłabienie mięśni występuje we wszystkich przypadkach ALS, ale nie zawsze musi być pierwszym objawem.
Wczesne objawy często obejmują niezdarność, nietypowe zmęczenie kończyn, skurcze mięśni oraz niewyraźną mowę. Objawy rozprzestrzeniają się na wszystkie części ciała w miarę postępu choroby.
Niektórzy pacjenci mogą mieć trudności z podejmowaniem decyzji oraz pamięcią, co ostatecznie prowadzi do postaci otępienia znanego jako demencja czołowo-czołowa.
Labilność emocjonalna może powodować wahania nastroju oraz nieprzewidywalne reakcje emocjonalne.
Leczenie i zapobieganie
Nie istnieje lekarstwo na ALS, dlatego leczenie koncentruje się na złagodzeniu objawów, zapobieganiu niepotrzebnym komplikacjom i spowolnieniu postępu choroby.
ALS prowadzi do szeregu zmian fizycznych, psychicznych i społecznych, dlatego często zespół specjalistów wspomaga pacjentów w radzeniu sobie z objawami i opieką, poprawiając ich jakość życia oraz przedłużając czas przeżycia.
Riluzol (Rilutek) został zatwierdzony przez Food and Drug Administration (FDA) w 1995 roku do leczenia ALS i wydaje się spowalniać postęp choroby. Może działać poprzez obniżenie poziomu glutaminianu w organizmie, ekscytotoksyny, która jest związana z uszkodzeniem neuronów.
W maju 2017 roku zatwierdzono lek Radicava (Edaravone) do leczenia ALS. Może spowolnić spadek sprawności fizycznej o jedną trzecią.
Wiele projektów badawczych ma na celu znalezienie sposobów wykorzystania nowych i istniejących leków w leczeniu różnych aspektów ALS. Lekarze mogą również przepisywać leki w celu leczenia różnych objawów.
Terapia
Fizjoterapia może pomóc pacjentom z ALS w opanowaniu bólu oraz w radzeniu sobie z problemami z poruszaniem się.
Fizjoterapeuta może zapewnić wsparcie oraz informacje:
- ćwiczenia o niskim wpływie, korzystne dla zdrowia sercowo-naczyniowego i ogólnego samopoczucia
- środki ułatwiające poruszanie się, takie jak chodziki i wózki inwalidzkie
- urządzenia ułatwiające życie, takie jak rampy
Terapia zajęciowa może wspierać pacjentów w utrzymaniu niezależności poprzez:
- pomoc w wyborze sprzętu adaptacyjnego i technologii wspomagających, co pozwala na zachowanie codziennej rutyny
- szkolenie w celu zrekompensowania osłabienia rąk i ramion
Z czasem może być konieczna terapia oddechowa, ponieważ mięśnie odpowiedzialne za oddychanie osłabiają się.
Urządzenia wspomagające oddychanie mogą pomóc pacjentowi w lepszym oddychaniu w nocy. Niektórzy pacjenci mogą potrzebować wentylacji mechanicznej, w której jeden koniec rurki jest podłączony do respiratora, a drugi wprowadzany jest do tchawicy przez chirurgicznie utworzony otwór w szyi (tracheostomię).
Terapia mowy jest pomocna, gdy ALS zaczyna utrudniać rozmowę. Terapeuci mowy mogą nauczyć pacjentów technik adaptacyjnych. Inne metody komunikacji obejmują pisanie oraz komputerowy sprzęt komunikacyjny.
Wsparcie żywieniowe jest kluczowe, ponieważ trudności z połykaniem mogą utrudnić uzyskanie wystarczającej ilości składników odżywczych. Specjaliści od żywienia mogą doradzić w zakresie przygotowywania pożywnych posiłków, które są łatwiejsze do połknięcia. Przydatne mogą być również przyssawki i rurki do karmienia.
Diagnoza
Nie istnieje jeden test, który mógłby zdiagnozować ALS, dlatego diagnoza opiera się na objawach i wynikach badań, które mają na celu wykluczenie innych stanów o podobnych objawach.
Testy, które mogą pomóc w diagnozowaniu ALS, to:
- elektromiografia (EMG), która wykrywa aktywność elektryczną w mięśniach
- Badanie przewodnictwa nerwowego (NCS), które ocenia, jak dobrze nerwy wysyłają sygnały
Testy te mogą pomóc w wykluczeniu neuropatii obwodowej, uszkodzenia nerwów obwodowych oraz miopatii (chorób mięśni).
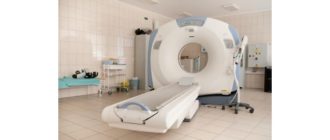
Skan MRI może ujawnić inne problemy, które mogą powodować objawy, takie jak guz rdzenia kręgowego czy przepuklina dysku w szyi.
Dodatkowe testy w celu wykluczenia innych stanów mogą obejmować badania krwi i moczu, a także biopsję mięśnia.
Problemy medyczne, które mogą wywoływać podobne objawy jak ALS, to HIV, choroba z Lyme, stwardnienie rozsiane (SM), wirus polio oraz wirus Zachodniego Nilu.
Jeśli występują objawy zarówno w górnym, jak i dolnym neuronie ruchowym, istnieje podejrzenie ALS.
Objawy górnego neuronu ruchowego obejmują sztywność, opór na ruchy mięśni oraz szybki refleks. Objawy dolnego neuronu ruchowego to osłabienie, atrofia mięśni oraz drżenie mięśni.
Wskazówki dotyczące życia z ALS
Kilka wskazówek może pomóc osobom z ALS oraz ich bliskim dostosować się do zmieniającej się sytuacji.
Bądź w kontakcie: kontakt społeczny jest niezwykle ważny. Pozostań w kontakcie z przyjaciółmi i staraj się uczestniczyć w jak największej liczbie wcześniejszych aktywności. Możesz znaleźć lokalne lub internetowe grupy wsparcia, które mogą odpowiedzieć na pytania i podzielić się doświadczeniami.
Bądź praktyczny: przygotuj torbę z chusteczkami, łatwymi w użyciu sztućcami oraz innymi niezbędnymi przedmiotami. Zarejestruj się, aby uzyskać tabliczkę z niepełnosprawnością dla samochodu. Dokonaj w domu regulacji, takich jak podnoszenie deski sedesowej.
Planuj z wyprzedzeniem: może być trudno, gdy bliska osoba nie może już wykonywać czynności, które wcześniej były dla niej proste. Przewidywanie potencjalnych ograniczeń może pomóc w przygotowaniu się na nadchodzące zmiany.
Poszukaj pomocy finansowej: leczenie ALS może być kosztowne. Dowiedz się, czy kwalifikujesz się do pomocy w ramach świadczeń z tytułu Ubezpieczeń Społecznych, Medicare, Medicaid lub Veteran Affairs.
Zadbaj o wsparcie dla opiekunów: umów się z przyjacielem lub członkiem rodziny, aby pomógł Ci w opiece nad osobą z ALS. Opiekunowie powinni również dbać o swoje zdrowie, aby móc lepiej wspierać swoich bliskich.
Twoje możliwości mogą być ograniczone przez sytuację finansową, ale lokalne grupy wsparcia mogą pomóc w radzeniu sobie z wyzwaniami finansowymi i emocjonalnymi związanymi z ALS, oferując porady lub praktyczną pomoc.
Nowe badania i odkrycia w 2024 roku
Warto zaznaczyć, że w 2024 roku prowadzone są intensywne badania nad ALS, które przynoszą nowe nadzieje dla pacjentów. W badaniach klinicznych testowane są nowe terapie genowe, które mogą potencjalnie spowolnić postęp choroby. Eksperci wskazują, że terapia komórkowa, w której wykorzystuje się komórki macierzyste, staje się obiecującym kierunkiem w walce z ALS.
Ponadto, niedawne badania pokazują, że czynniki takie jak dieta, styl życia, a nawet regularna aktywność fizyczna mogą mieć pozytywny wpływ na przebieg choroby. Utrzymanie zdrowej diety bogatej w antyoksydanty oraz unikanie substancji toksycznych mogą przyczynić się do poprawy jakości życia pacjentów. Wzrost zainteresowania terapiami wspomagającymi, takimi jak akupunktura czy medytacja, również może przynieść ulgę w objawach i poprawić samopoczucie.
Dzięki współpracy naukowców, lekarzy oraz organizacji pacjentów, w przyszłości możemy spodziewać się przełomowych odkryć, które przyczynią się do lepszego zrozumienia oraz leczenia stwardnienia zanikowego bocznego.