Choroby neuronów ruchowych to grupa stanów, które prowadzą do stopniowej utraty funkcji nerwów w mózgu i rdzeniu kręgowym. Choć są rzadkie, stanowią poważne i nieuleczalne formy postępującej neurodegeneracji.
Neurony ruchowe, odpowiedzialne za przekazywanie sygnałów elektrycznych do mięśni, odgrywają kluczową rolę w funkcjonowaniu organizmu, a ich uszkodzenie wpływa na zdolność ruchową.
Choroba neuronów ruchowych (MND) może wystąpić w każdym wieku, jednak najczęściej diagnozowana jest u osób powyżej 40. roku życia, a mężczyźni są bardziej narażeni niż kobiety.
Najczęstszy typ, stwardnienie zanikowe boczne (ALS), dotyka około 30 000 Amerykanów w danym momencie, a rocznie stawia się ponad 5000 nowych diagnoz.
Znany fizyk Stephen Hawking żył z ALS przez wiele dekad, aż do swojej śmierci w marcu 2018 roku. Wirtuoz gitary Jason Becker to kolejny znany przypadek osoby, która od lat zmaga się z ALS.
Szybkie fakty dotyczące chorób neuronów ruchowych
Oto kilka kluczowych punktów dotyczących chorób neuronów ruchowych. Więcej szczegółów znajduje się w głównym artykule.
- Choroby neuronów ruchowych (MND) to grupa stanów, które wpływają na komórki nerwowe, które wysyłają wiadomości do mózgu.
- Występuje stopniowe osłabienie wszystkich mięśni w ciele, co ostatecznie wpływa na zdolność oddychania.
- Genetyka, wirusy i czynniki środowiskowe mogą odgrywać rolę w rozwoju MND.
- Nie ma lekarstwa, ale terapie wspomagające mogą poprawić jakość życia.
- Średnia długość życia po rozpoznaniu waha się od 3 do ponad 10 lat.
Rodzaje

Istnieje kilka rodzajów chorób neuronu ruchowego.
ALS, czyli choroba Lou Gehriga, jest najczęstszym typem, wpływającym na mięśnie rąk, nóg, ust oraz układu oddechowego. Średni czas przeżycia wynosi od 3 do 5 lat, ale niektórzy pacjenci, korzystając z odpowiedniej opieki, żyją nawet 10 lat lub dłużej.
Postępujące porażenie opuszkowe (PBP) obejmuje pień mózgu i często towarzyszy ALS. Powoduje problemy z mówieniem, połykaniem oraz częste zadławienia.
Postępująca atrofia mięśniowa (PMA) prowadzi do stopniowego zaniku mięśni, szczególnie w ramionach, nogach i ustach, i może być wariantem ALS.
Pierwotna stwardnienie boczne (PLS) to rzadki typ MND, który postępuje wolniej niż ALS. Choć nie jest śmiertelny, znacząco wpływa na jakość życia. U dzieci występuje młodzieńcze pierwotne stwardnienie boczne.
Zanik mięśniowy kręgosłupa (SMA) to dziedziczna forma MND, która dotyka dzieci. Istnieją trzy typy, wszystkie spowodowane przez nieprawidłowy gen znany jako SMA1. Wpływają głównie na tułów, nogi i ramiona. Perspektywy długoterminowe różnią się w zależności od rodzaju.
Różne typy MND mają podobne objawy, ale postępują z różnymi prędkościami i różnią się nasileniem.
Objawy
MND można podzielić na trzy etapy: wczesny, średni i zaawansowany.
Objawy i oznaki wczesnego stadium
Objawy rozwijają się powoli i mogą być mylone z innymi chorobami neurologicznymi.
Wczesne objawy zależą od tego, który układ ciała jest dotknięty jako pierwszy. Typowe objawy pojawiają się w jednym z trzech obszarów: rąk i nóg, ust (opuszki) lub układu oddechowego.
Zawierają:
- osłabienie chwytu, utrudniające chwytanie i utrzymywanie przedmiotów
- zmęczenie
- bóle mięśniowe, skurcze i drżenia
- niewyraźna, a czasem zniekształcona mowa
- osłabienie rąk i nóg
- zwiększona niezdarność i potknięcia
- trudności z połykaniem
- trudności w oddychaniu lub duszność
Znaki i objawy na etapie średnim

W miarę postępu choroby objawy stają się bardziej intensywne.
- Ból i osłabienie mięśni rosną, a skurcze i wzdęcia stają się bardziej dokuczliwe.
- Kończyny stopniowo tracą swoją siłę.
- Mięśnie kończyn mogą ulegać kurczeniu.
- Ruch w dotkniętych kończynach staje się coraz trudniejszy.
- Mięśnie kończyn mogą stać się nienormalnie sztywne.
- Ból stawów wzrasta.
- Jedzenie, picie i połykanie stają się trudniejsze.
- Nadmierne wydzielanie śliny może wystąpić z powodu trudności w kontrolowaniu śliny.
- Ziewanie może występować w sposób niekontrolowany.
- Ból szczęki może wynikać z nadmiernego ziewania.
- Problemy z mową nasilają się, gdy mięśnie w gardle i ustach słabną.
Osoba może wykazywać zmiany w osobowości oraz stanie emocjonalnym, z epizodami niekontrolowanego płaczu lub śmiechu.
W przeszłości sądzono, że MND nie wpływa znacząco na funkcjonowanie mózgu ani pamięć, jednak badania wskazują, że aż 50 procent pacjentów z ALS doświadcza pewnych problemów z funkcjami poznawczymi.
Mogą one obejmować trudności z pamięcią, planowaniem, językiem, zachowaniem oraz orientacją przestrzenną. Do 15 procent pacjentów z ALS może mieć postać demencji znaną jako demencja czołowo-skroniowa (FTD).
Problemy z oddychaniem mogą wystąpić, gdy przepona, główny mięsień oddechowy, zaczyna zawodzić. Możliwe jest wystąpienie duszności, nawet podczas snu. Ostatecznie pacjenci mogą potrzebować wsparcia w oddychaniu.
Wtórne objawy to bezsenność, lęk i depresja.
Zaawansowane objawy i oznaki
Ostatecznie pacjent staje się całkowicie zależny od innych, nie jest w stanie poruszać się, jeść ani oddychać bez wsparcia. Bez opieki wspierającej, osoba ta umrze. Pomimo najlepszej dostępnej opieki, powikłania układu oddechowego pozostają najczęstszą przyczyną zgonów.
Przyczyny
Neurony ruchowe wysyłają sygnały z mózgu do mięśni i kości, co umożliwia ruchy mięśni. Są zaangażowane zarówno w ruchy świadome, jak i automatyczne, takie jak połykanie i oddychanie.
Niektóre przypadki MND mają charakter dziedziczny, inne występują sporadycznie. Dokładne przyczyny pozostają niejasne, jednak National Institute of Neurological Diseases and Stroke (NINDS) sugeruje, że czynniki genetyczne, toksyczne, wirusowe oraz inne aspekty środowiskowe mogą mieć istotne znaczenie.
Czynniki ryzyka
Oto niektóre z czynników ryzyka związanych z MND:
Dziedziczność: w Stanach Zjednoczonych około 1 na 10 przypadków ALS jest dziedziczonych. SMA jest również znane jako dziedziczny warunek.
Wiek: po 40 roku życia ryzyko znacząco wzrasta, chociaż pozostaje stosunkowo niskie. ALS najczęściej pojawia się w wieku od 55 do 75 lat.
Płeć: Mężczyźni są bardziej narażeni na rozwój MND.
Niektórzy eksperci sugerują, że doświadczenia wojskowe mogą zwiększać ryzyko rozwoju choroby.
Badania sugerują, że zawodowi piłkarze są bardziej narażeni na śmierć z powodu ALS, choroby Alzheimera oraz innych chorób neurodegeneracyjnych w porównaniu do ogółu społeczeństwa, co może wskazywać na związek z nawracającymi urazami głowy oraz chorobami neurologicznymi.
Diagnoza

Wczesne stadium MND może być trudne do zdiagnozowania, ponieważ objawy są podobne do tych występujących w innych schorzeniach, takich jak stwardnienie rozsiane (MS), zapalenie nerwu czy choroba Parkinsona.
Zazwyczaj lekarz pierwszego kontaktu kieruje pacjenta do neurologa, specjalisty w zakresie diagnostyki i leczenia chorób układu nerwowego.
Neurolog zaczyna od dokładnego zebrania wywiadu i przeprowadzenia badania fizykalnego układu nerwowego.
Dodatkowe testy mogą być również pomocne.
Badania krwi i moczu: Analizy te mogą wykluczyć inne stany oraz wykryć wzrost poziomu kinazy kreatynowej, co może wskazywać na uszkodzenie mięśni.
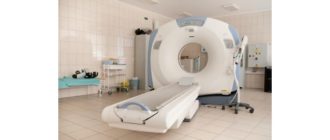
Skan MRI: Choć nie może wykryć MND, może pomóc wykluczyć inne schorzenia, takie jak udar, guz mózgu czy problemy z krążeniem mózgowym.
Elektromiografia (EMG) oraz badanie przewodnictwa nerwowego (NCS): Te testy są często stosowane razem. EMG ocenia aktywność elektryczną w mięśniach, podczas gdy NCS bada prędkość przewodzenia sygnałów elektrycznych w mięśniach.
Nakłucie lędźwiowe: Pozwala na analizę płynu mózgowo-rdzeniowego, który otacza mózg i rdzeń kręgowy.
Biopsja mięśnia: Jeśli lekarz podejrzewa chorobę mięśniową zamiast MND, może zlecić biopsję mięśnia.
Po przeprowadzeniu testów lekarz zazwyczaj obserwuje pacjenta przez pewien czas, zanim postawi diagnozę MND.
Kryteria znane jako kryteria El Escorial mogą pomóc lekarzowi w ocenie charakterystycznych objawów neurologicznych, które mogą wspierać rozpoznanie ALS.
Kryteria te obejmują:
- kurczenie się mięśni, osłabienie lub drganie
- sztywność mięśni lub nieprawidłowe odruchy
- rozprzestrzenianie się objawów na nowe grupy mięśni
- brak innych czynników wyjaśniających objawy
Leczenie

Obecnie nie ma lekarstwa na MND, dlatego leczenie koncentruje się na spowolnieniu postępu choroby oraz maksymalizacji niezależności i komfortu pacjenta.
Terapie mogą obejmować korzystanie z urządzeń wspomagających oddychanie, karmienie, mobilność oraz komunikację.
Rehabilitacja może obejmować terapię fizyczną, zajęciową oraz logopedyczną.
Spowolnienie postępu choroby
Dwa leki zostały zatwierdzone przez FDA do leczenia ALS. Riluzol (Rilutek) zmniejsza poziom glutaminianu w organizmie, co wydaje się być najskuteczniejsze we wczesnych stadiach choroby oraz u osób starszych. Wykazano, że poprawia przeżywalność.
W 2017 roku zatwierdzono lek Radicava (Endaravone) do leczenia ALS. Jego dokładny mechanizm działania nie jest w pełni poznany, ale może opóźniać postęp choroby poprzez działanie przeciw uszkodzeniu tkanek.
Naukowcy prowadzą badania nad możliwością zastosowania komórek macierzystych w terapii ALS.
Skurcze mięśni i sztywność
Skurcze mięśni oraz sztywność można leczyć za pomocą fizykoterapii i leków, takich jak iniekcje toksyny botulinowej (BTA), które blokują sygnały od mózgu do mięśni przez około 3 miesiące.
Baklofen, środek rozluźniający mięśnie, może zmniejszyć sztywność. Wprowadza się go za pomocą małej pompy, która jest chirurgicznie wszczepiana i połączona z przestrzenią wokół rdzenia kręgowego.
Baklofen blokuje niektóre sygnały nerwowe odpowiedzialne za spastyczność i może pomóc w przypadku nadmiernego ziewania.
Leczenie ślinotoków
Skopolamina, stosowana w terapii choroby lokomocyjnej, może pomóc w kontrolowaniu objawów ślinotoków i jest aplikowana w formie plastrów za uchem.
Niekontrolowany śmiech lub płacz
Leki przeciwdepresyjne, zwane inhibitorami wychwytu zwrotnego serotoniny (SSRI), mogą pomóc w epizodach niekontrolowanego śmiechu lub płaczu, znanych jako niestabilność emocjonalna.
Mowa, terapia zajęciowa i fizyczna
Pacjenci z trudnościami w mowie mogą nauczyć się użytecznych technik od wykwalifikowanego terapeuty mowy. W miarę postępu choroby pacjenci często potrzebują wsparcia w komunikacji.
Terapia fizyczna i zajęciowa wspierają mobilność oraz funkcjonowanie, a także pomagają w redukcji stresu.
Trudności z połykaniem (dysfagia)
Gdy jedzenie i picie stają się trudniejsze, pacjent może potrzebować przezskórnej endoskopowej gastrostomii (PEG), czyli rurki do karmienia umieszczonej w brzuchu, co jest stosunkowo prostą procedurą.
Ból
Niesteroidowe leki przeciwzapalne (NSAID), takie jak ibuprofen, mogą łagodzić ból związany z skurczami mięśni. W zaawansowanych stadiach choroby, leki takie jak morfina mogą pomóc w łagodzeniu bólu stawów i mięśni.
Problemy z oddychaniem
Mięśnie oddechowe osłabiają się stopniowo, ale czasami może wystąpić nagłe pogorszenie.
Wentylacja mechaniczna może być niezbędna w przypadku trudności w oddychaniu. Maszyna pobiera powietrze, filtruje je i pompuje do płuc, często przez tracheostomię, czyli chirurgiczny otwór w szyi, który umożliwia wspomagane oddychanie.
Niektórzy pacjenci korzystają z terapii uzupełniających, w tym specjalnych diet bogatych w witaminy. Chociaż MND nie można wyleczyć, zdrowa dieta może poprawić ogólny stan zdrowia i samopoczucie.
Przeszczep komórek macierzystych w leczeniu ALS
Badania nad komórkami macierzystymi oraz terapią genową wydają się obiecujące w leczeniu ALS w przyszłości, lecz potrzebne są dalsze badania.
Perspektywy
MND zazwyczaj prowadzi do śmierci. W zależności od rodzaju, większość osób nie przetrwa dłużej niż pięć lat po pojawieniu się objawów, chociaż niektórzy pacjenci żyją 10 lat lub dłużej, w zależności od stopnia i tempa progresji choroby.
Narodowa Służba Zdrowia w Wielkiej Brytanii (NHS) zauważa, że życie z MND może być niezwykle trudne, a diagnoza często wydaje się «przerażająca».
Jednak z odpowiednim wsparciem społeczności oraz bliskich, jakość życia pacjentów często jest lepsza niż oczekiwano, a zaawansowany etap «zwykle nie jest nieprzyjemny», co dla większości osób oznacza spokojne odejście w domu, podczas snu.
Pacjenci mogą chcieć przygotować dyrektywę na wypadek, gdyby nie mogli wyrazić swoich życzeń dotyczących leczenia. Może to obejmować miejsce, w którym pragną być leczeni w końcowych etapach, na przykład w szpitalu, w ośrodku hospicyjnym lub w domu z opieką hospicyjną, a także decyzje dotyczące mechanicznego wspomagania oddychania czy innej pomocy.
W miarę trwania badań naukowych, naukowcy mają nadzieję na lepsze zrozumienie MND i poszukiwanie nowych metod leczenia.