Talasemia to dziedziczna choroba krwi, która znacząco wpływa na zdolność organizmu do produkcji hemoglobiny oraz czerwonych krwinek. Osoby z talasemią mają zbyt mało czerwonych krwinek oraz hemoglobiny, co skutkuje ich mniejszym rozmiarem. Wpływ schorzenia może wahać się od łagodnego do ciężkiego, a w niektórych przypadkach zagraża nawet życiu.
Co roku, na całym świecie, diagnozuje się około 100 000 noworodków z poważnymi postaciami talasemii. Najczęściej występuje ona u osób z przodkami z rejonów basenu Morza Śródziemnego, Azji Południowej oraz Afryki.
Objawy
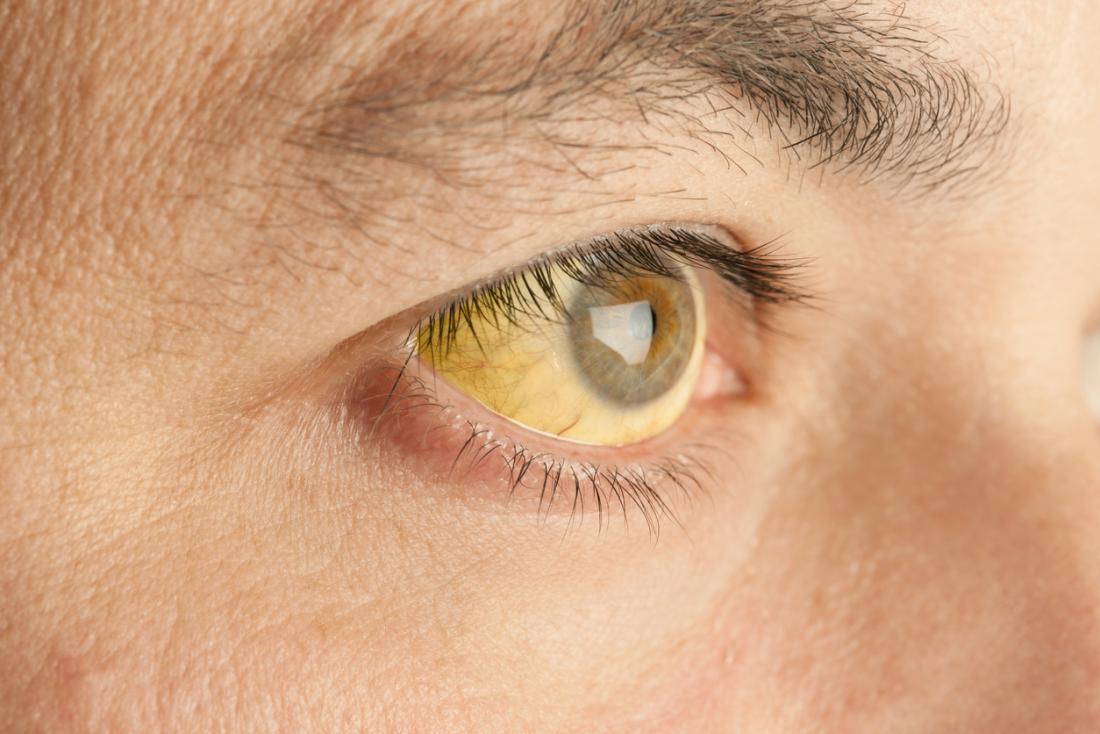
Objawy talasemii są zróżnicowane i zależą od jej rodzaju. U większości niemowląt z beta-talasemią oraz niektórymi typami talasemii alfa, objawy mogą być niewidoczne do ukończenia 6. miesiąca życia. Dzieje się tak, ponieważ noworodki produkują hemoglobinę płodową, która później jest zastępowana przez «normalną» hemoglobinę, co może prowadzić do pojawienia się symptomów.
Objawy talasemii mogą obejmować:
- żółtaczkę oraz bladość skóry
- senność i chroniczne zmęczenie
- ból w klatce piersiowej
- zimne dłonie i stopy
- duszność
- kurcze nóg
- szybkie bicie serca
- słabe karmienie u niemowląt
- opóźniony wzrost
- bóle głowy
- zawroty głowy i omdlenia
- większą podatność na infekcje
Dodatkowo, deformacje szkieletu mogą pojawić się, gdy organizm stara się produkować więcej szpiku kostnego. W przypadku nadmiaru żelaza, organizm może próbować wchłonąć jeszcze więcej żelaza, co z kolei może prowadzić do uszkodzeń wątroby, serca oraz śledziony.
Pacjenci z hemoglobiną H są bardziej narażeni na rozwój kamieni żółciowych oraz powiększenie śledziony. Bez odpowiedniego leczenia, powikłania talasemii mogą prowadzić do niewydolności narządów.
Leczenie
Leczenie talasemii zależy od jej rodzaju oraz ciężkości. Obejmuje ono:
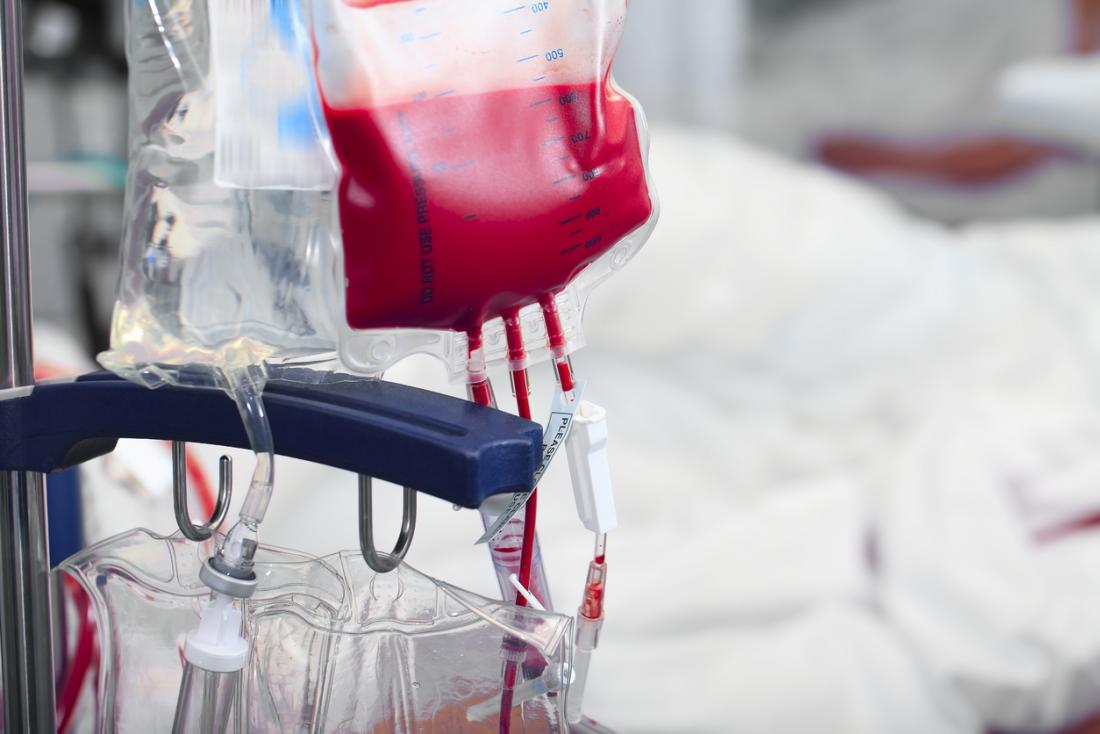
1. **Transfuzje krwi:** Są one kluczowe w uzupełnieniu poziomu hemoglobiny i czerwonych krwinek. Pacjenci z ciężką talasemią mogą potrzebować od ośmiu do dwunastu transfuzji rocznie, podczas gdy ci z łagodnymi postaciami mogą wymagać do ośmiu transfuzji rocznie, zwłaszcza w okresach stresu czy infekcji.
2. **Chelatacja żelaza:** To proces usuwania nadmiaru żelaza z organizmu, co jest istotne, ponieważ transfuzje krwi mogą prowadzić do przeładowania żelazem, szkodliwego dla serca i innych narządów. Pacjenci mogą stosować deferoksaminę lub deferazyrok.
3. **Pobranie komórek macierzystych lub przeszczep szpiku kostnego:** Jest to skuteczna metoda leczenia w ciężkich przypadkach, pozwalająca na produkcję zdrowych krwinek.
4. **Operacje:** Mogą być konieczne w celu skorygowania deformacji kości.
5. **Terapia genowa:** Naukowcy prowadzą badania nad zastosowaniem technik genetycznych do leczenia talasemii, w tym wprowadzanie normalnych genów beta-globiny do szpiku kostnego pacjenta.
Przyczyny
Hemoglobina jest białkiem transportującym tlen w organizmie, a jej produkcja wymaga żelaza, które pozyskujemy z pożywienia. U osób z talasemią szpik kostny nie wytwarza wystarczającej ilości zdrowej hemoglobiny lub czerwonych krwinek, co prowadzi do niedotlenienia organizmu i anemii.
Osoby z łagodną talasemią mogą nie wymagać leczenia, natomiast cięższe formy wymagają regularnych transfuzji krwi.
Diagnoza

Większość dzieci z umiarkowaną lub ciężką talasemią jest diagnozowana do ukończenia 2. roku życia. Osoby bez objawów nie mają często świadomości, że są nosicielami, dopóki nie urodzą dziecka z talasemią.
Badania krwi mogą wykazać, czy dana osoba jest nosicielem lub ma talasemię. Kluczowe testy to:
- Pełne badanie krwi (CBC): Sprawdza poziom hemoglobiny oraz liczbę i wielkość czerwonych krwinek.
- Liczba retikulocytów: Mierzy szybkość produkcji czerwonych krwinek przez szpik kostny.
- Badania żelaza: Pomagają określić przyczynę anemii.
- Testy genetyczne: Analiza DNA wykaże obecność mutacji genów.
- Badania prenatalne: Umożliwiają ocenę, czy płód ma talasemię.
- Pobieranie próbek kosmówki (CVS): Fragment łożyska jest pobierany do badania.
- Amniopunkcja: Niewielka próbka płynu owodniowego jest analizowana.
Rodzaje
Hemoglobina składa się z czterech łańcuchów alfa-globiny i dwóch beta-globiny. Wyróżniamy dwa główne rodzaje talasemii: alfa i beta.
Talassemia alfa
W alfa-talasemii organizm nie produkuje wystarczającej ilości białka alfa. Potrzebne są cztery geny, po dwa na każdym chromosomie 16. Brak jednego lub więcej genów prowadzi do rozwoju talasemii alfa. Nasilenie objawów zależy od liczby uszkodzonych genów.
– **Jeden wadliwy gen:** Pacjent nie wykazuje objawów, jest nosicielem.
– **Dwa wadliwe geny:** Pacjent ma łagodną anemię (alfa talasemia minor).
– **Trzy wadliwe geny:** Pacjent cierpi na chorobę hemoglobiny H, wymaga regularnych transfuzji.
– **Cztery wadliwe geny:** Główna talasemia alfa, najcięższa postać, zwykle prowadzi do obrzęków płodu.
Beta talasemia
Beta talasemia występuje, gdy występują mutacje w genach beta-globiny. Ciężkość choroby zależy od liczby uszkodzonych genów:
– **Jeden wadliwy gen:** Beta-talasemia.
– **Dwa wadliwe geny:** Mogą występować umiarkowane lub ciężkie objawy (beta talasemia major).
Beta talasemia jest częstsza wśród ludzi o śródziemnomorskich korzeniach, a jej występowanie jest wyższe w Afryce Północnej oraz Azji Zachodniej.
Komplikacje
Talasemia może prowadzić do wielu komplikacji zdrowotnych.
Przeciążenie żelaza
Częste transfuzje krwi mogą prowadzić do nadmiaru żelaza, co zwiększa ryzyko zapalenia wątroby, zwłóknienia i marskości wątroby. Uszkodzenia przysadki mózgowej mogą prowadzić do opóźnionego dojrzewania, a w późniejszym wieku do cukrzycy oraz problemów z tarczycą.
Alloimmunizacja
Czasami transfuzja krwi wywołuje reakcję immunologiczną, co podkreśla znaczenie dokładnego dopasowania grupy krwi.
Powiększona śledziona
W talasemii krwinki czerwone często mają nieprawidłowy kształt, co utrudnia ich przetwarzanie przez śledzionę, prowadząc do jej powiększenia oraz nadmiernej aktywności.
Infekcje
Usunięcie śledziony zwiększa ryzyko infekcji, a transfuzje krwi mogą prowadzić do zakażeń chorobami przenoszonymi przez krew.
Deformacje kości
Rozszerzenie szpiku kostnego może prowadzić do deformacji kości, zwłaszcza czaszki i twarzy, co zwiększa ryzyko złamań.
Życie z talasemią
Osoby z talasemią mogą potrzebować stałej opieki medycznej, aby skutecznie zarządzać chorobą. Kluczowe jest przestrzeganie planu transfuzji oraz chelatacji.

Osoby z talasemią powinny:
- uczestniczyć w regularnych wizytach kontrolnych
- utrzymywać kontakt z bliskimi oraz grupami wsparcia
- przestrzegać zdrowej, zróżnicowanej diety
- zapewnić sobie odpowiednią ilość aktywności fizycznej
Należy unikać niektórych produktów, takich jak szpinak czy zboża wzbogacone w żelazo, aby zapobiec nadmiernemu gromadzeniu się tego pierwiastka. Warto omówić z lekarzem kwestie diety oraz aktywności fizycznej.
Centra Kontroli i Zapobiegania Chorobom (CDC) zalecają regularne aktualizowanie szczepień, aby zapobiegać chorobom, co jest szczególnie istotne dla pacjentów po transfuzjach krwi.
Talasemia a ciąża
Osoby rozważające ciążę powinny skonsultować się z genetykiem, szczególnie jeśli oboje partnerzy są nosicielami talasemii. Kobiety w ciąży z talasemią mogą mieć wyższe ryzyko kardiomiopatii oraz cukrzycy, a także mogą wystąpić ograniczenia wzrostu płodu.
Matki z beta-moll talasemią powinny być pod stałą opieką kardiologa lub hematologa, aby minimalizować potencjalne problemy zdrowotne.
Perspektywy
Perspektywy dla pacjentów z talasemią zależą od jej rodzaju. Osoby z cechą talasemii mogą prowadzić normalne życie, jednak powikłania związane z beta-talasemią mogą prowadzić do przedwczesnej śmierci, zwłaszcza przed 30. rokiem życia.